
Structural, Energetic, and Electronic Properties of H-Interstitial in C-Monovacancy: A First-Principles Density Functional Theory
DOI:
https://doi.org/10.48048/tis.2024.7657Keywords:
C-monovacancy, H-interstitial, Formation energy, Reaction coordinate, Band structureAbstract
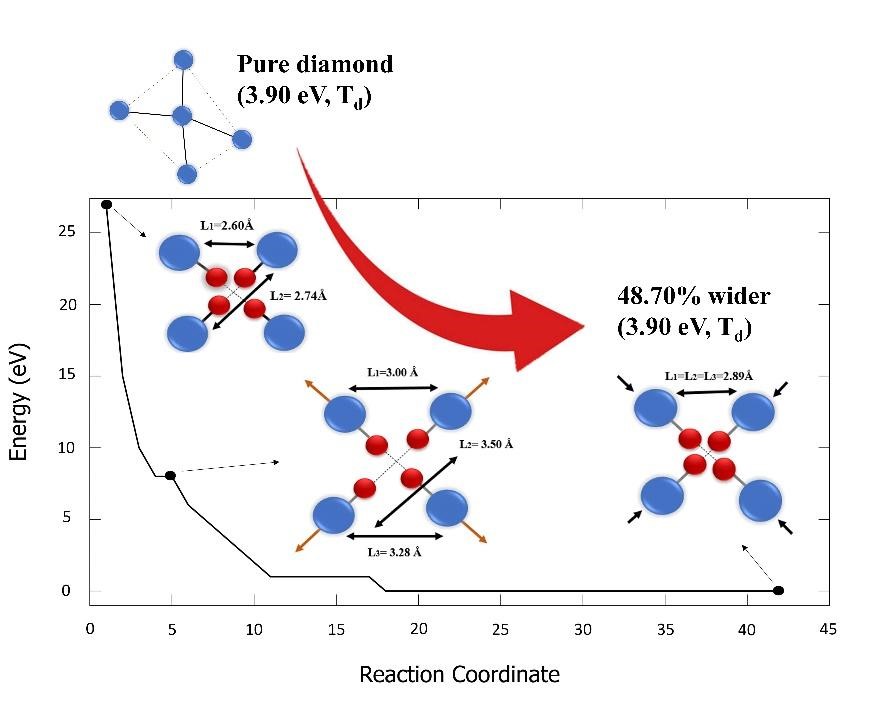
We discover a unique structural-modified diamond that exhibits similar symmetry and band gap energy to that of the pure diamond. We study a complex carbon-vacancy-hydrogen in the diamond using the density-functional-theory method. The defective models are created by adding H-interstitial (Hi, where i = 1, 2, 3 and 4) in the 3D diamond C-monovacancy. The result shows that carbon-vacancy-hydrogen defects significantly decreased the symmetry from Td to C2V. Likewise, the volumetric size of the systems is widening up to 48.70 %, while the optimized band gap energies are narrowing. Additional states appeared in the C-monovacancy, H1-V, H2-V, and H3-V systems which further improved electron mobility. The Hi compensates the C-monovacancy which further serves as a deep donor. Interestingly, H4-V exhibits similar symmetry and band gap energy to that of the pure diamond, but its volumetric size is 48.70 % wider.
HIGHLIGHTS
- Study of complex carbon-vacancy-hydrogen defects through density-functional-theory calculations
- Discover a unique structural-modified diamond that is 48.70 % wider than the pure diamond, but exhibits similar symmetry and band gap energy.
- Complex carbon-vacancy-hydrogen defects significantly lower the symmetries, widening the volumetric sizes, and narrowing the band gap energies.
- The H-interstitial acts as a deep donor.
GRAPHICAL ABSTRACT

Downloads
References
A Chepurov, V Sonin, D Shcheglov, E Zhimulev, S Sitnikov, A Yelisseyev and A Chepurov. Surface Porosity of Natural Diamond Crystals after the Catalytic Hydrogenation. Crystals 2021; 11, 1341.
S Łoś, K Fabisiak, K Paprocki, M Szybowicz, A Dychalska, E Spychaj-Fabisiak and W Franków. The hydrogenation impact on electronic properties of p-diamond/n-Si heterojunctions. Materials 2021; 14, 6616.
H Gomez, MN Groves and MR Neupane. Study of the structural phase transition in diamond (100) & (111) surfaces. Carbon Trends 2011; 3, 33.
ZS Fatomi, AD Nugraheni and Sholihun. Vibrational effect on vacancy concentration in diamond: The density-functional-theory calculation. Comput. Condens. Matter 2022; 32, e00708.
Y Dai, CX Yan, AY Li, Y Zhang and SH Han. Effects of hydrogen on electronic properties of doped diamond. Carbon 2005; 43, 1009-14.
CJH Wort and RS Balmer. Diamond as an electronic material. Mater. Today 2008; 11, 22-8.
J Isberg, J Hammersberg, E Johansson, T Wikström, DJ Twitchen, AJ Whitehead, SE Coe and GA Scarsbrook. High carrier mobility in single-crystal plasma-deposited diamond. Science 2002; 297, 1670-2.
H Sakaue, N Yoshimura, S Shingubara and T Takahagi. Low dielectric constant porous diamond films formed by diamond nanoparticles. Appl. Phys. Lett. 2003; 83, 2226-8.
JP Goss, R Jones, MI Heggies, CP Ewels, PR Briddon and S Öberg. Theory of hydrogen in diamond. Phys. Rev. B 2002; 65, 115207.
D Purnawati, N Fajariah, H Prayogi, JP Bermundo, AD Nugraheni and Sholihun. Dissociation-energy calculations of C-multivacancies in diamond: The density-functional-theory study. Jpn. J. Appl. Phys. 2023; 62, 051002.
XJ Hu, YB Dai, RB Li, HS Shen and XC He. The diffusion of vacancies near a diamond (001) surface. Solid State Comm. 2002; 122, 45-8.
B Slepetz and M Kertesz. Divacancies in diamond: A stepwise formation mechanism. Phys. Chem. Chem. Phys. 2014; 16, 1515-21.
R Jones, LS Hounsome, N Fujita, S Öberg and PR Briddon. Electrical and optical properties of multivacancy centres in diamond. Phys. Status Solidi 2007; 204, 3059-64.
DP Hastuti, W Amalia, Z Priska, P Nurwantoro and Sholihun. First-principles density-functional-theory calculations of formation and dissociation energies in germanene multivacancies. Mater. Today Comm. 2020; 22, 100754.
M Saito, K Yamashita and T Oda. Magic numbers of graphene multivacancies. Jpn. J. Appl. Phys. 2007; 46, L118.
DP Hastuti, P Nurwantoro and Sholihun. Stability study of germanene vacancies: The first-principles calculations. Mater. Today Comm. 2018; 19, 459-63.
X Chang, Q Xue, D He, L Zhu, X Li and B Tao. 585 divacancy-defective germanene as a hydrogen separation membrane: A DFT study. Int. J. Hydrogen Energ. 2017; 42, 24189-96.
S Lebègue and O Eriksson. Electronic structure of two-dimensional crystals from ab initio theory. Phys. Rev. B 2009; 79, 115409.
K Takeda and K Shiraishi. Theoretical possibility of stage corrugation in Si and Ge analogs of graphite. Phys. Rev. B 1994; 50, 14916.
T Yamasaki, A Kuroda, T Kato, J Nara, J Koga, T Uda, K Minami and T Ohno. Multi-axis decomposition of density functional program for strong scaling up to 82,944 nodes on the K computer: Compactly folded 3D-FFT communicators in the 6D torus network. Comput. Phys. Comm. 2019; 244, 264-76.
Sholihun, W Amalia, DP Hastuti, P Nurwantoro, AD Nugraheni and RHS Budhi. Magic vacancy-numbers in h-BN multivacancies: The first-principles study. Mater. Today Commun. 2019; 20, 100591.
A Abdurrazaq, AT Raji and WE Meyer. Effect of isovalent doping on hydrogen passivated vacancy-oxygen defect complexes in silicon: Insight from density functional theory. Silicon 2021; 13, 1969-77.
JP Goss. Theory of hydrogen in diamond. J. Phys. Condens. Matter. 2003; 15, R551.
P Briddon, R Jones and GMS Lister. Hydrogen in diamond. J. Phys. C Solid State Phys. 1988; 21, L1027.
D Saada, J Adler and R Kalish. Lowest-energy site for hydrogen in diamond. Phys. Rev. B 2000; 61, 10711.
W Amalia, P Nurwantoro and Sholihun. Density-functional-theory calculations of structural and electronic properties of vacancies in monolayer hexagonal boron nitride (h-BN). Comput. Condens. Matter 2019; 16, e00354.
F Birch. Finite elastic strain of cubic crystals. Phys. Rev. 1947; 71, 809.
AK McMahan. Interstitial-sphere linear muffin-tin orbital structural calculations for C and Si. Phys. Rev. B 1984; 30, 5835.
MT Yin and ML Cohen. Ground-state properties of diamond. Phys. Rev. B 1981; 24, 6121.
W Kaiser and WL Bond. Nitrogen is a major impurity in common type I diamonds. Phys. Rev. 1959; 115, 857.
T Yamanaka, S Morimoto and H Kanda. Influence of the isotope ratio on the lattice constant of diamond. Phys. Rev. B 1994; 14, 9341.
H Holloway, KC Hass, MA Tamor, TR Anthony and WF Banholzer. Isotopic dependence of the lattice constant of diamond. Phys. Rev. B 1991; 13, 7123.
D Richards and M Amos. Shape optimization with surface-mapped CPPNs. IEEE Trans. Evol. Comput. 2016; 21, 391-407.
S Sholihun, HP Kadarisman and P Nurwantoro. Density-functional-theory calculations of formation energy of the nitrogen-doped diamond. Indonesian J. Chem. 2018; 18, 749-54.
Z Priska, S Hidayati, S Sholihun, W Amalia and P Nurwantoro. Hydrogen and water adsorptions on monolayer hexagonal boron nitride (h-BN): The first-principles calculations. Key Eng. Mater. 2021; 884, 387-93.
YN Apriati, AD Nugraheni and S Sholihun. Interaction of C60 with small molecules: Adsorption - inclusion energy calculation using the density functional theory. Mater. Sci. Forum 2022; 1066, 135-43.
C Glover, ME Newton, PM Martineau, S Quinn and DJ Twitchen. Hydrogen incorporation in diamond: The vacancy-hydrogen complex. Phys. Rev. Lett. 2004; 92, 135502.
DEP Vanpoucke and K Haenen. Revisiting the neutral C-vacancy in diamond: Localization of electrons through DFT+ U. Diam. Relat. Mater. 2017; 79, 60-9.
A Zelferino, S Salustro, J Baima, V Lacivita, R Orlando and R Dovesi. The electronic states of the neutral vacancy in diamond: A quantum mechanical approach. Theor. Chem. Accounts Theor. Comput. Model. Theor. Chim. Acta 2016; 135, 74.
AA Emery and C Wolverton. High-throughput DFT calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci. Data 2017; 4, 170153.
RQ Hood, PRC Kent, RJ Needs and PR Briddon. Quantum monte Carlo study of the optical and diffusive properties of the vacancy defect in diamond. Phys. Rev. Lett. 2003; 91, 076403.
K Ramakrishna and J Vorberger. Ab-initio dielectric response function of diamond and other relevant high-pressure phases of carbon. J. Phys. Condens. Matter 2019; 32, 095401.
P Deák, B Aradi, T Frauenheim, E Janzén and A Gali. Accurate defect levels were obtained from the HSE06 range-separated hybrid functional. Phys. Rev. B 2010; 81, 153203.
F Tran and P Blaha. Importance of the kinetic energy density for band gap calculations in solids with density functional theory. J. Phys. Chem. 2017; 121, 3318-25.
O Madelung. Semiconductors-basic data. Springer Book Archive. Springer Berlin, Heidelberg, Germany, 1996.
SP Gao. Band gaps and dielectric functions of cubic and hexagonal diamond polytypes calculated by many-body perturbation theory. Phys. Status Solidi B 2015; 252, 235-42.
Y Ma, J Ma, Y Lv, J Liao, Y Ji and H Bai. Effect of mono vacancy defect on the charge carrier mobility of carbon nanotubes: A case study on (10,0) tube from first-principles. Superlattice. Microst. 2016; 99, 140-4.
Downloads
Published
How to Cite
Issue
Section
License
Copyright (c) 2023 Walailak University

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.



